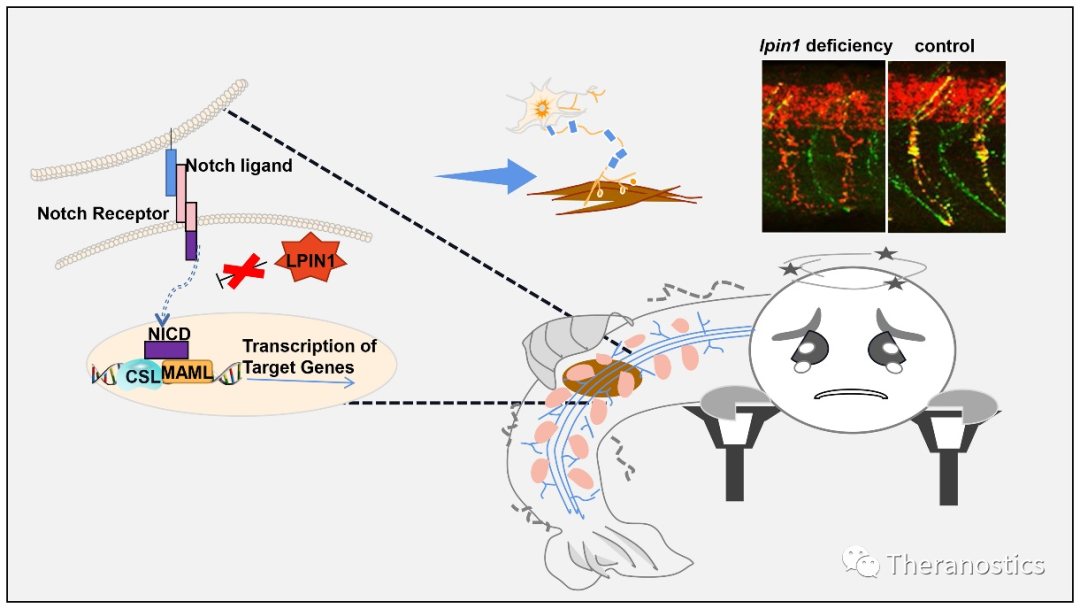
肌无力是由肌肉、神经和代谢紊乱引起的一种症状,可在任何年龄和种族背景人群发病。肌萎缩、炎性肌病、神经源性肌病、运动神经元疾病和重症肌无力是常见的由遗传原因导致的肌无力相关疾病。导致肌无力的潜在分子过程和遗传原因十分复杂,需要研究人员不断探索。近日,威廉希尔官网田静教授为首的研究团队成员在生物医学1区杂志《Theranostics》(2020 IF: 8.579)上发表论文,首次发现人类LPIN1的复合杂合突变会导致成年发病肌无力综合征伴周围神经病变。研究运用了斑马鱼模型对致病机理进行深入分析,该成果将加深我们对LPIN1突变引起人类疾病的理解,拓宽LPIN1在神经肌肉形成中的作用,对人类肌无力致病机制和治疗途径的探究具有一定指导意义。
Lipin 1又称磷脂酸磷酸水解酶1(PAP1),是复发性肌红蛋白尿(MIM#268200)的致病基因,在体内具有双向调控脂质代谢的功能。本研究首次报道了LPIN1基因两个新的突变位点:c.2047A>C(p.I683L) 和c.2201G>A(p.R734Q),该复合杂合突变会导致成人肌无力综合征并伴随有神经功能障碍,但病人无肌红蛋白尿表征。通过斑马鱼模型体内模拟lpin1基因缺陷在机体发育和神经发生过程中的发病机制,发现 lpin1缺失会影响肌块发育完整性,抑制初级运动神经元和次级运动神经元萌发,导致突触后乙酰胆碱受体定位异常,以及髓鞘形成缺陷,最终导致斑马鱼接触反应障碍和游泳行为异常,部分模拟人类LPIN1突变患者肌无力表型。
斑马鱼lipin 1功能缺失导致的神经肌肉表型被发现与异常上调的Notch信号有关。为研究这一调控机制的保守性,团队利用LPIN1突变细胞系,在人胚肾细胞(HEK293T)、胶质母细胞瘤细胞(U87)、小鼠成肌细胞(C2C12)和人骨肉瘤细胞(U2OS)的实验中表明,LPIN1通过NOTCH信号通路调节神经肌肉的发育和功能。
以上研究结果首次发现人类LPIN1突变会导致成人肌无力综合征并伴随肌纤维萎缩和神经脱髓鞘,揭示了lipin 1调控神经肌肉发育的重要分子机制。
威廉希尔官网田静教授和闽江大学陈建明教授为文章的共同通讯作者,威廉希尔官网博士研究生卢淑娴和吕赵劼为文章并列第一作者。
参考文献:
Shuxian Lu, Zhaojie Lyu, Zhihao Wang, Yao Kou, Cong Liu, Shengyue Li, Mengyan Hu, Hongjie Zhu, WenxingWang, Ce Zhang, Yung-Shu Kuan, Yi-Wen Liu, Jianming Chen, Jing Tian. Lipin 1deficiency causes adult-onset myasthenia with motor neuron dysfunction in humans and neuromuscular junction defects in zebrafish. Theranostics. 2021;11(6): 2788-2805.
原文链接:https://www.thno.org/v11p2788.htm